固相微萃取GC-MS/MS方法应用
固相微萃取GC-MS/MS方法应用
固相微萃取GC-MS/MS方法快速定量分析食品和饮料中被限制的具有生物学活性的调味剂
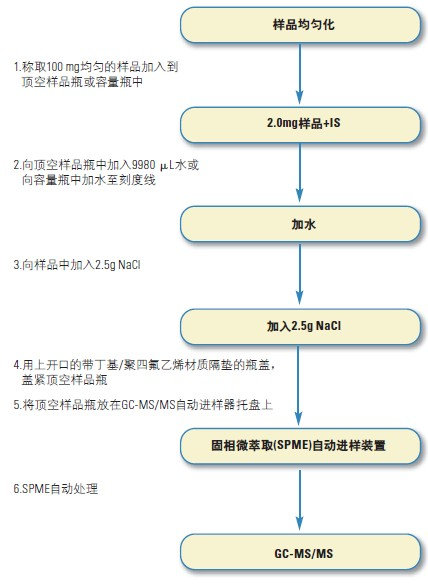
1.分析流程图
2.引言
在食品和饮料行业中,尽管植物提取物具有非常久远的使用历史,欧盟对于某些植物提取物进行了管制2,如黄樟油精,在美国其被禁止直接作为食品添加剂使用3。欧盟1334/2008法规2禁止了15种化学纯的调味剂被直接作为食品或饮料添加剂使用。这些调味品包括:琼脂酸、芦荟苷(芦荟素)、辣椒素、香豆素、海棠素、β-细辛脑、草蒿脑(甲基胡椒酚)、氢氰酸、薄荷呋喃、甲基丁子香酚、胡薄荷酮、苦木素、黄樟油精、石蚕苷A、α和 β-侧柏酮。只有在调味品和食物中本身天然存在、且在规定的浓度水平以下时,食品和饮料中可允许存在上述15物种质中的10种物质2。就物理性质和化学组成而言,该规定涉及的食品种类非常繁多,例如,液体食品(烈酒和不含酒精的饮料)、半固体食品(汤羹、酱汁和甜点)以及固体食品(甜食类、口香糖、鱼类、肉类、烘烤食品和早餐谷类)。然而,目前食品行业中尚无针对这些物质可用的常规分析方法,而使得很难对加工完成后的食品中这些调味剂物质的最终浓度进行有效控制。特别是,在不同植物种类中这些调味剂的含量差异非常大,也使食品加工中很难控制其最终含量。对挥发性物质的分析,顶空分析法是一种非常有前景的分析方法。这是因为该方法仅需最简单的样品制备过程,并可以实现自动化分析。在顶空分析法中,相对于其他的顶空分析技术,固相微萃取(SPME)具有很多优点,而在食品分析中可能是目前应用最为广泛的一种分析方法4。尽管针对调味剂的SPME分析方法已经非常完善了,但是目前发表的方法都只对单一类别的食品进行分析。而对于具有生物学活性的调味剂的执行强制管理控制,尚未发表可真正提供解决方案的方法。本文介绍了一种SPME方法,对于三种类型的食品,利用优化的通用方法,可同时测定7种挥发性调味剂的含量,而欧盟2对特定食品中的这些调味剂浓度均有规定。
3.适用范围
本方法可用于含酒精和不含酒精的饮料、半固体加工的食品和固体食品中含量为0.5至3000 mg/kg的7种具有生物学活性的调味剂的定性和定量检测。这些调味剂包括:香豆素、β-细辛脑、草蒿脑(甲基胡椒酚)、薄荷呋喃、甲基丁子香酚、胡薄荷酮和侧柏酮。
4.分析原理
本方法利用自动顶空固相微萃取(HS/SPME)方法,使用聚二甲基硅氧烷(PDMS)SPME纤维,可从各种基质中萃取目标化合物(即,具有生活学活性的调味剂)。将外加了内标化合物、水和氯化钠的样品置于顶空样品瓶中。将顶空样品瓶密封,待其顶部空间达到平衡后,利用Thermo ScientificTSQ Quantum XLS气相色谱-三重四极杆质谱仪系统对样品瓶顶部空间同时自动采样和分析。
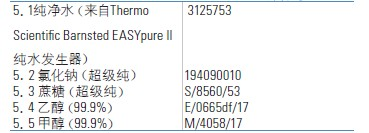
5.试剂清单
6.校正标准物质
6.1 生物学活性调味剂
6.1.1 β-细辛脑,纯度72%(Ehrenstorfer博士提供)
6.1.2 香豆素(1,2-苯并吡喃酮),纯度99.5%(Ehrenstorfer博
士提供)
6.1.3 草蒿脑(1-烯丙基-4-甲氧基苯),纯度_98.5%(Sigma-
Aldrich)
6.1.4 薄荷呋喃,纯度_98.5%(Sigma-Aldrich)
6.1.5 甲基丁子香酚(4-烯丙基-1,2-二甲氧基苯),纯度99.5%
(Sigma-Aldrich)
6.1.6 胡薄荷酮,纯度98.8%(Sigma-Aldrich)
6.1.7 侧柏酮(α和β),纯度_99%(Sigma-Aldrich)
6.2 内标化合物
6.2.1 香豆素(5,6,7,8-D4),浓度为100 μg/mL的丙酮溶液
(Ehrenstorfer博士提供)
6.1.2 二环己基甲醇,纯度98%(Sigma-Aldrich)
7. 标准物质制备
7.1 调味剂的标准储备液(1000 μg/mL):称取25.00 mg调味剂化合物(根据标准物质的实际纯度重新计算用量)加入容量瓶中,用甲醇溶解并稀释至25 mL。该溶液可在4°C至少保存3个月。
7.2 七种调味剂的工作标准溶液(1 μg/mL,对香豆素浓度为10 μg/mL):分别移取25 μL 浓度为1000 μg/mL的侧柏酮、薄荷呋喃、草蒿脑、胡薄荷酮、甲基丁子香酚和β-细辛脑储备液,和250 μL 浓度为1000 μg/mL的香豆素储备液于25 mL容量瓶中,加入水稀释至刻度线(得到10 μg/mL的香豆素工作液;其他6种为1 μg/mL)。工作标准溶液需要每次使用前现配。
7.3 内标化合物二环己基甲醇的标准储备液(1000 μg/mL):称取25.00 mg化合物(根据标准物质的实际纯度重新计算用量)加入容量瓶中,用甲醇溶解并稀释至25mL。该溶液可在4°C至少保存3个月。
7.4 内标化合物二环己基甲醇的工作标准溶液(10 μg/mL):移取100 μL 浓度为1000 μg/mL的二环己基甲醇的标准储备液至10 mL容量瓶中,加入水稀释至刻度线。工作标准溶液需要每次使用前现配。
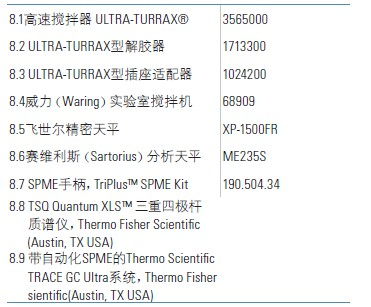
8.仪器
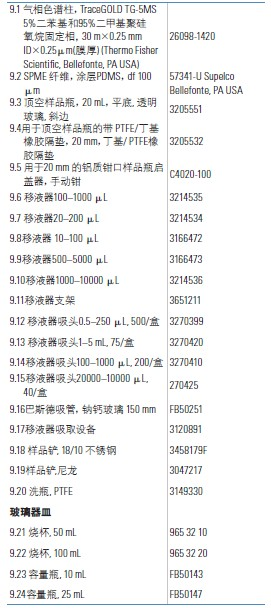
9. 消耗品
10.分析流程
仪器准备:在使用仪器前或准备用SPME模式进行分析工作前,请仔细阅读Thermo Scientific TriPlus仪器操作指南的相关章节和Thermo Scientific TriPlus标准操作流程的第IV部分内容。其描述了在SPME手柄和萃取头安装过程中所有必需的维护注意事项。
10.1 样品制备
固体或半固体基质样品
10.1.1 将150 g样品在高速搅拌机(适用于汤羹、酱汁、蒜酱)或威力实验搅拌机(适用于固体基质,如牛奶什锦早餐)中搅拌5 min,得到均匀的样品。然后准确称取0.1 g 3样品直接放入顶空样品瓶中。
10.1.2 使用合适的微量移液器分别移取10 μL二己基甲醇的工作标准溶液、10 μL香豆素-d4的标准溶液和9980 μL水加入顶空样品瓶中。液体基质样品
10.1.3称取0.1 g样品直接放入顶空样品瓶中,并使用合适的
微量移液器移取10 μL二己基甲醇的工作标准溶液、10μL香豆素-d4的标准溶液和9980 μL水加入顶空样品瓶中。
10.1.4 上述两种情况下都需要加入2.5 g NaCl, 并使用带PTFE/丁基橡胶隔垫的样品盖和样品钳盖紧样品瓶。
10.1.5使用下列空白食品代表性每类基质进行空白校正。
•液体基质样品(主要指酒类)的空白样品:40 %的乙醇水溶液。
•半固体基质样品(主要指酱汁和蒜酱)的空白样品:纯番茄酱汁。
•固体基质样品(主要指牛奶什锦早餐)的空白样品:燕麦粥。
•对于固体和半固体基质样品,标准溶液和内标样品的
用量见表1,对于液体基质样品其对应的标准溶液和内标样品用量见表2。
10.2 SPME自动化分析
10.2.1 使用涂覆100 μm的聚二甲基硅氧烷纤维(PDMS-100),并按照生产厂商推荐的方法将纤维插入GC进样器,预处理备用。
10.2.2将含测试样品的顶空样品瓶放置在样品托盘中(最大可放置54 个样品瓶),并加载SPME自动进样器。
10.2.3 开始SPME分析程序。其操作程序为:在50℃条件下将样品旋转(液体形成漩涡)5 min,将纤维头插入样品瓶的顶部空间,并在溶液漩涡条件下于50℃萃取40min;然后将纤维转移至进样口并于250 ℃脱附5 min。该程序结束后,将纤维头转移到第二个进样口(取代老化平台)进行清洗并于250 ℃老化处理5 min。
10.3 GC分析
GC分析工作在带自动SPME分析系统的TRACE GC Ultra™仪器系统上进行(Thermo Fisher Scientific, Austin, TX USA)。GC分析条件为:
:TraceGOLD TG-5MS, 5% 二苯基和95%二甲基聚硅氧烷为固定相(30 m长,内径0.25 mm,膜厚0.25μm)
进样模式:不分流
进样口温度:250 ℃
左侧载气流量:1.2 mL/min
分流量:50 mL/min
不分流时间:3 min
老化进样口温度:250℃
右侧载气流量:0.1 mL/min
传输管线温度:250℃
炉温:60℃保持1 min, 以15℃/min加热到120℃,保持2 min;
再以30℃/min加热至280℃保持10 min。
10.4 串联MS/MS检测
使用TSQ Quantum XLS三重四极杆质谱仪(Thermo FisherScientific, Austin, TX USA)进行质谱分析。
离子化模式:电子碰撞(EI)正离子,电离能70 eV发射电流:30 μA
离子源温度:250℃
扫描方式:选择性反应监测(SRM)
循环时间:0.1 s
峰宽:Q1/Q3 半峰高宽(FWHM)为0.70 Da
碰撞气体(Ar)压力:1.0 mTorr
用于目标化合物和内标化合物的选择性反应监测分析的参
数见表3。
11. 结果计算
11.1定性分析
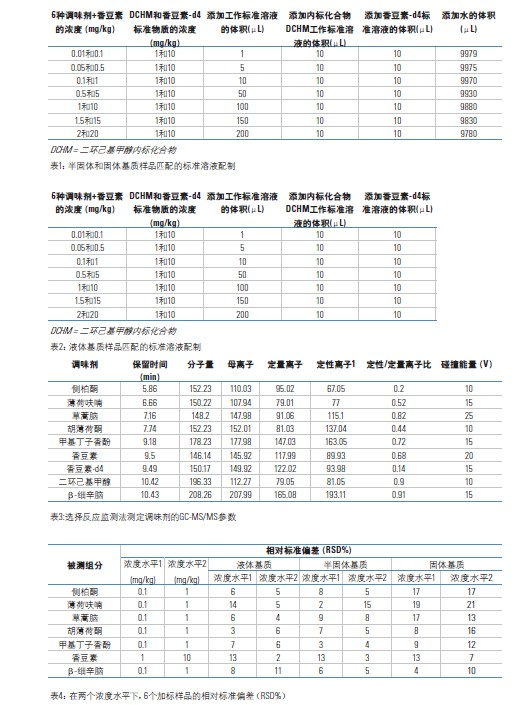
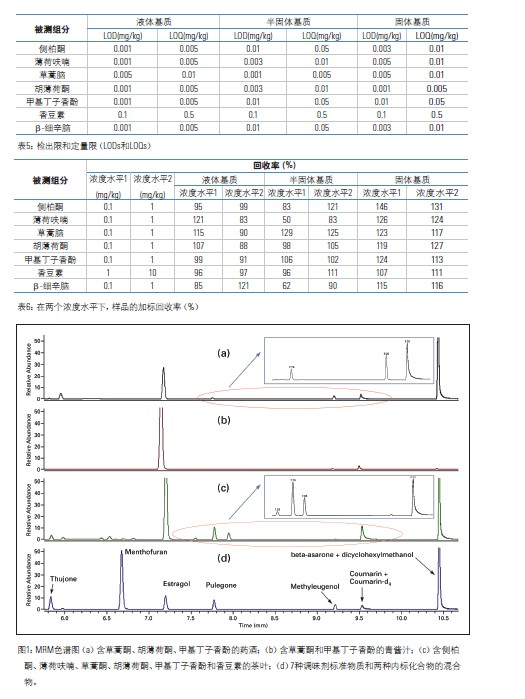
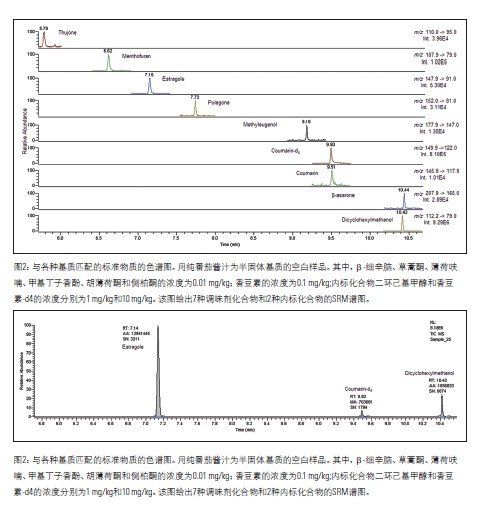
根据对应标准样品的保留时间(±0.05%),可确定离子对离子(定性和定量离子)的存在。在多重反应监测(MRM)模式中,测得的样品中定性离子对定量离子的峰面积之比应当接近(±20%)于标准样品中的定性离子对定量离子的峰面积之比,可参阅表3。定性离子和定量离子,是根据所选择的母离子产生的子离子的峰强度来选择的。
11.2 定量分析
本方法使用与每种基质匹配的内标化合物的峰面积比进行定量分析。二己基甲醇被用作其中6种调味品化合物(即,侧柏酮、薄荷呋喃、草蒿酮、胡薄荷酮、甲基丁子香酚和β-细辛脑)的内标化合物。香豆素-d4被用作香豆素的内标化合物。根据被测物质对内标化合物的相对峰面积对被测化合物的浓度作校正曲线。样品中调味剂的浓度(cf)由下式进行计算:

12. 方法验证
根据方法的特异性、线性、精密度、检出限(LOD)和定量限(LOQ)、准确度和适应性验证了该方法。最后,经证实,本方法可以很好地应用于检测大量商业化食品中的目标调味剂。通过在空白基质(固体样品:燕麦粥;半固体样品:纯番茄酱汁;液体样品:乙醇水溶液)中加入目标化合物的加标实验测试了该方法的性能。
12.1 特异性
使用选择性反应监测(SRM)方法,通过与对应的标准样品对比,样品中离子对离子(定性和定量)的色谱保留时间是否在正确的时间出现,确认了该方法的特异性。如表3所示,该方法实际测得样品中定性离子对定量离子的峰面积之比与标准样品中的定性离子对定量离子的峰面积之比接近。
12.2 线性和校正曲线
对于6种调味剂在0.01–2.0 mg/kg和香豆素在0.1–2.0 mg/kg范围之间评价了校正曲线的线性。所有情况下,线性相关系数都大于0.99。将7种与基质匹配的校正标准物质重复添加到每个批次的试验样品中,进而得到了校正曲线。
12.3 精密度
通过在三种不同基质中添加两个浓度水平的(内标)样品,分别重复测定6次得到该方法的相对标准偏差(%RSD)。对于液体基质样品,选择乙醇水溶液(40%)作为空白基质(并添加不同含量的蔗糖,以模拟酒类和能量型饮料);对半固体基质样品,选择番茄酱汁作为空白基质;对固体基质样品则选择燕麦粥为空白基质样品。添加样品的浓度为0.1和1 mg/kg,每个样品重复分析6次。对6种调味剂(β-细辛脑、草蒿酮、薄荷呋喃、甲基丁子香酚、胡薄荷酮和侧柏酮),添加的第一个浓度水平为0.1 mg/kg,而添加的香豆素的浓度为1 mg/kg。对6种调味剂的第二个浓度水平为1 mg/kg,而对香豆素仍然选择较高的浓度水平10 mg/kg。表4给出了该方法的精密度测定结果,相对标准偏差(RSDs)在2-21%之间。由此可知,该方法对液态和半液态食品的精密度优于固态食品,但是所有精密度结果对于(质量)监管分析方法而言都是可以接受的。
12.4检出限(LOD)和定量限(LOQ)
按照IUPAC方法,估算了该方法的检出限和定量限。即分析空白样品获得该方法的噪声水平,进而用信噪比的3倍和10倍分别代表LODs和LOQs。表5列出了该方法对于三种不同基质(固体、半固体和液体基质)的LODs和LOQs。对于所有的情况,该方法的LODs和LOQs值都优于食品管理限值中规定的0.5 mg/kg的最低控制水平。
12.5 准确度
通过比较实验测定的样品浓度和添加的标准物质的实际浓度评价了该方法的准确度。使用优化的方法分析了三种基质样品。用加标的40%乙醇水溶液代表液体基质样品,加标的番茄酱汁代表半固体基质样品,加标的燕麦粥代表固体基质样品。在6次重复试验中,添加了0.1和1 mg/kg的2个浓度水平的内标。对6种调味剂(β-细辛脑、草蒿酮、薄荷呋喃、甲基丁子香酚、胡薄荷酮和侧柏酮),添加内标的第一个浓度水平为0.1 mg/kg,而添加的香豆素的浓度为1 mg/kg。对6种调味剂,添加的第二个浓度水平为1 mg/kg,而对香豆素仍然选择较高的浓度水平10 mg/kg。如表6所示,除了固体基质样品为正误差外,该方法均具有良好的准确度。
13.结论
单一的 分析结果确认了该方法能用于复杂食品中有含量限制的七种具有生物学活性的调味剂含量检测。基于所选择的液体、半固体或固体基质类别的通用方法和每种类型食品对应的优化的分析测试条件,该方法可适用于所有类型的食品中具有生物学活性的调味剂的含量分析。该方法具有足够高的灵敏度,可满足食品行业对调味剂含量的质量监管的需求;使用MS/MS检测器,基于检测离子比,可保证该方法高度可靠的、准确的对化合物进行定性鉴定。因此,我们推荐该方法用于食品行业中具有生物学活性的调味剂的强制限值检测分析。
14. References 5
1. For details of this research please see: Bousova K., Mittendorf
K., Paez V.,Senyuva H., A Solid-Phase Micro-Extraction GCMS/
MS Method for Rapid Quantitative Analysis of Food and
Beverages for the Presence of Legally Restricted Biologically
Active Flavorings. J. AOAC. Accepted for publication, 2011.
2. Regulation (EC) No 1334/2008 of 16 December 2008 on
flavorings and certain food ingredients with flavoring properties
for use in and on foods and amending Council Regulation (EEC)
No 1601/91, Regulations (EC) No 2232/96 and (EC) No 110/2008
and Directive 2000/13/EC. Official Journal of the European
Union. (2008) L 354/34-50.
3. Code of Federal Regulations Title 21CFR189.180 [Revised as of
April 1, 2010]
4. Kataoka, H., Lord, H. L., & Pawliszyn, J. (2000). J. Chromatogr.,
A, 880, 35-62.
展源
何发
相关文章
-

AAS法分析茶叶中的铅,镉,砷
2020-05-27
-

三聚氰胺,你还要害多少人
2020-05-27
-
红外光谱分析,你了解多少?
2021-01-11
-

QC, IQC, IPQC, QA,到底是什么鬼?
2020-05-27
-

选对 ,快速开发方法
2020-05-27
-

做PGS/MS分析时,如何防止焦油状污染物进入毛细柱
2020-05-27
-

HPLC检测器,你了解吗?
2024-03-06
-

做PGS/MS分析时,如何防止焦油状污染物进入毛细柱?
2020-05-27
-

超净工作台原理,使用与维护
2020-05-27

加载更多