解决峰拖尾与峰前延小妙招!

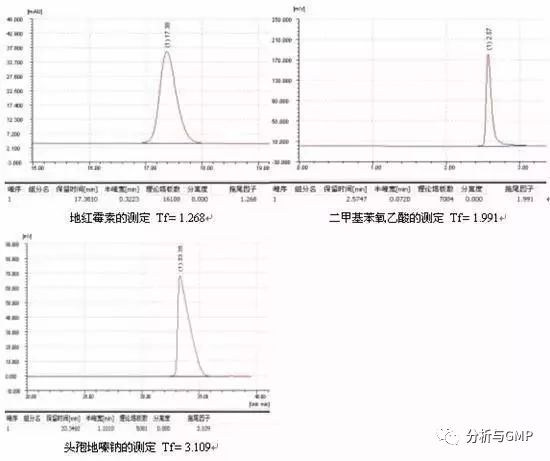
反相色谱中,通常非极性和弱极性的化合物能获得良好的峰形,而带有-COOH、-NH2、-NHR、-NR2等极性基团的化合物则比较容易产生拖尾,原因是硅胶表面残留硅羟基对极性和碱性样品分子产生次级保留效应。
反相填料表面有残余的硅羟基,具有一定的酸性,其pKa约为4.5~4.7。根据电离规律,流动相的pH-pKa>2即pH>6.7时,99%以上的硅羟基应该是呈离子状态的,即Si-O- ,而pKa-pH>2即pH<2.5时,酸性环境抑制了硅羟基的电离,99%以上的硅羟基应该是呈分子状态的,即Si-OH,但其极性仍然存在,即Si-Oδ-Hδ+。Si-Oδ-Hδ+和-Si-O-对于极性化合物之间的作用力则是一种极性的静电作用力,这种作用力比范德华力要强得多。同时,因为硅胶表面键合了C18长链,由于空间位阻作用,样品分子中能接触到残余硅羟基的机会不多,只有少部分的分子才能与残余硅羟基产生强的静电作用而被推迟洗脱出来,产生后拖。拖尾严重的程度与样品分子极性大小和残余硅羟基的多少有着直接的关系。
完全硅羟化的硅胶表面硅羟基浓度为8mol/m2,由于空间位阻的作用,通常与C18硅烷基发生反应的仅达2~3.5mol/m2,需要再用小分子的硅烷跟硅羟基反应,如图中的三甲基氯硅烷,使硅羟基中的氢变成了三甲基硅烷的一个惰性基团。三甲基硅烷还是比氢原子大得多,还是不能将所有的残余硅羟基的活性封闭掉。
相同的样品在不同品牌的柱子上产生拖尾的严重性不同,从填料合成的角度而言就是键合密度是否高、封尾是否彻底,高密键合和彻底的封尾能显著减少这种机会,获得良好的峰形。
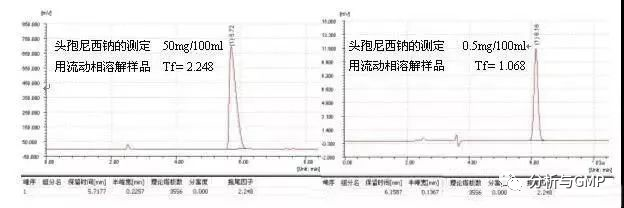
由于样品过载引起的峰形后拖不能通过改变色谱条件消除。如果发现过载,需降低进样量(包括进样体积或浓度),进样量越小,峰形越好,例子如头孢尼西钠的测定:
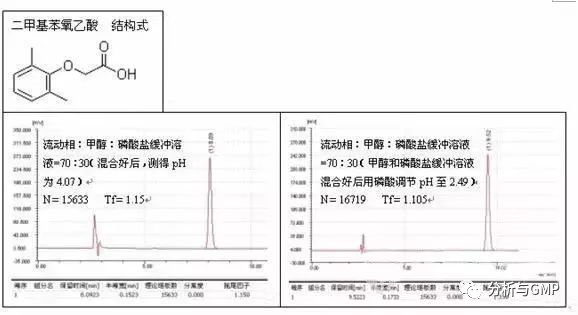
2)调节流动相pH
将流动相的pH调至比弱酸pKa小2以上,比弱碱pKa大2以上,可有效抑制易离子化待测物的电离,从而取得良好峰形。例子如二甲基苯氧乙酸的测定:
碱性化合物中,甲胺的pKa为10.64,那么在pH< 8.64的时候它是完全呈离子态的, 那么pH在2.5~8.64时,特别是pH6.7~8.64时,此时甲胺和硅羟基均以离子状态-NH3+和Si-O-形式存在的,它们之间相互吸引的静电作用导致后拖,这就是为什么很多碱性化合物在pH 7.0的条件下容易产生拖尾的原因。而很多 生产商也因此以阿米替林(含-N(CH3)2,碱性比-NH2强)在pH 7.0的条件下来评价和比较不同 之间的优劣,该条件下的阿米替林的峰形越好说明封尾越好。而在pH<2.5的条件下,也仍然后拖,是因为-NH3+足够强,即使硅羟基以分子状态存在也仍能够与Si-Oδ-Hδ+中氧原子产生吸引作用,导致峰形拖尾。当pH>12.64,大于pKa2以上时,甲胺完全以分子状态形式存在,不会引起拖尾。
3)增加缓冲盐或增大缓冲盐的浓度
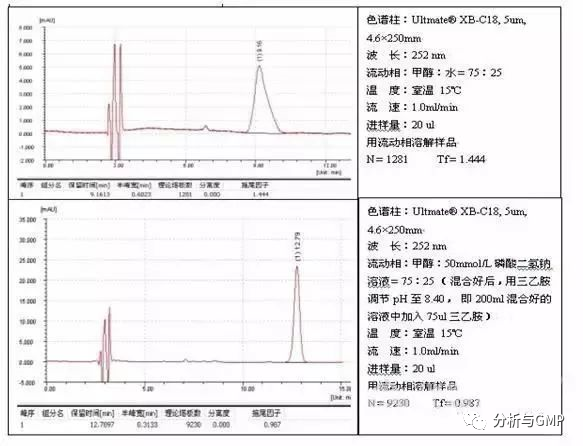
流动相中加入缓冲盐,增强了流动相的离子强度,在-NH3+等极性基团和硅羟基Si-O-之间存在着许多离子的干扰,减少了样品分子与硅羟基之间相互接触的机会,有助于削弱极性基团与硅羟基之间的相互作用,改善峰形。这种适合于碱性较弱(如氨基的N原子与强吸电子基相连),或碱性很强,但在刚性结构中,比较难以接近被C18长链和封尾试剂覆盖的硅羟基,例子如高乌甲素的测定:
4)加入三乙胺、四丁基硫酸氢铵或辛烷磺酸钠等
拖尾的产生是因为-NH2、-NHR、-NR2与硅羟基发生静电作用引起的,那么阻碍它们之间静电作用的途径,应该有两种,一种是占据硅羟基这个作用位点,另一种是占据-NH2、-NHR、-NR2这个作用位点。
在流动相中加入三乙胺,能显著的改善峰形,消除拖尾,其作用是屏蔽硅羟基。三乙胺的pKa为11.09,在pH<11.09-2=9.09的条件下是以离子状态N+H(CH2CH3)3的形式存在的,因此pH在2.5~8.64 硅羟基呈Si-O-离子态的情况下,N+H(CH2CH3)3容易与Si-O-形成相对较强的静电吸引力。
加入三乙胺改善峰形的时候,有两点需要注意:
1)三乙胺的碱性很强,加入三乙胺后流动相的pH可能超出 的使用范围,对 造成损伤。pH的改变也会导致出峰时间的显著变化,可能引起的其它问题,建议流动相中加入三乙胺后回调至未加入前的pH值。通常即使pH回调过后,由于三乙胺成为了固定相的一部分,保留时间也有较大变化;
2)三乙胺在210nm处有比较强的吸收,如果液相方法中检测波长在210nm以下测定时需要谨慎使用。
四烷基季铵盐(如四丁基硫酸氢铵、四丁基溴化铵、四丁基氢氧化铵等)在水中电离后,也形成了类似N+H(CH2CH3)3的结构N+(CH2CH2CH2CH3)4 ,这种结构也能有效的与Si-O-产生较强的静电作用,阻止氨基与硅羟基的接触,改善峰形。而且它还有一个不同于三乙胺的显著特点是,它在低波长范围的吸收比三乙胺弱。

峰前延发生的概率比较小,下面是三种前拖峰的表现形式:
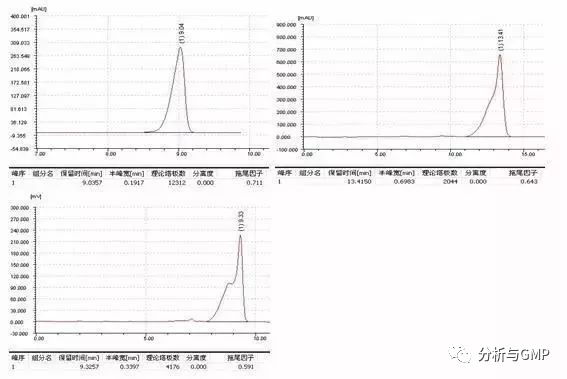
造成前延峰的原因有多种,我们可依次逐一排查:
1)样品是否过载
降低进样浓度,看峰形是否有所改善。一般认为峰高在100mAU左右比较合适,不至于因过载影响峰形。
2) 检查是否是用流动相溶解样品
溶解样品的溶剂(如纯甲醇)洗脱能力比流动相强会发生峰前延。
具体机理是:正常的峰形应该是样品在 上均匀的前移的情况下得到的,浓度分布在整个通过 柱床的过程中任何时候都呈正态分布。样品溶液进样后到达 时间很短,应还未被流动相充分稀释,洗脱能力更强的样品溶剂的局部存在,将使部分样品被洗脱的速度加快,导致峰前延。
3)增加流动相中缓冲盐的浓度
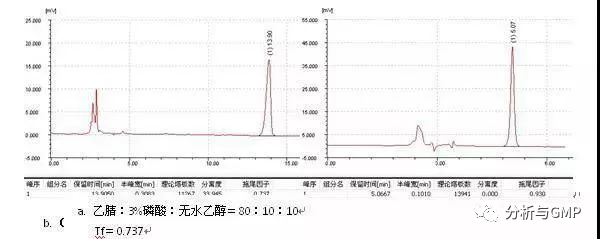
往流动相中加入少量的四氢呋喃有时可以改善峰形、增大分离度,很多色谱工作者都知道和使用,但其机理似乎少人提及。通常所加入的量在5%以内即可,需要的时候可以加入更大的量。
4)升高柱温
升温有助于增加流动相传质速率,减少因静电作用引起的前拖,但温度不宜太高,温度太高容易损伤 ,特别是含有离子对试剂的时候,最好不要超过40度。
柱头污染和柱头塌陷都会造成裂峰,最常见的原因是筛板上颗粒物堵塞样品进入 不均一,只需反冲 将颗粒物除掉就可解决。
1. 保留时间的重现性
在色谱分析中最重要的是分离选择性,保留时间的重现性则是可简单通过用峰面积定量代替峰高定量来消除其对分析结果的影响。温度、流动相组成、pH、离子强度等的微小变化都会引起保留时间的改变。10-20柱体积流动相平衡是必须的。三乙胺等添加剂加入,平衡时间需加长。
固定相流失:
1) 在酸性条件下键合相碳链水解断裂流失;
2)碱性条件下硅胶基质溶解导致在基质上键合固定相流失;
3)pH2-8下,上述两种因素导致的流失仍会缓慢发生。多位键合比单点键合好,长链比短链键合牢固,用于封尾的键合相易流失;C18链的稳定性比CN基链大三个数量级,一个是3个月会有明显流失,一个是1小时;用有机相洗柱子会加快流动相的流失。
柱污染:污染物作为固定相的一部分起保留作用,随着污染程度的逐步增加,保留时间发生趋向性的变化。
流动相组成变化: 如甲醇等挥发引起保留时间逐步变大。
相塌陷:纯水条件下,C18柱会发生固定相塌陷,引起保留时间逐步变小甚至失去保留能力。
2. 柱与柱之间的重现性
同一批填料生产出的不同柱子,装柱密度或柱体积的不同引起保留时间不一致。同样尺寸规格柱子,填料装得越结实,柱体积越小,保留时间越快。正常情况只有百分之几的差别。
平衡程度、老化程度、使用历史及污染程度的不一样都会导致两根同品牌同批次 之间的保留时间不一致。
3. 批与批之间的重现性
由于不同批次填料之间的差异性导致。主要是表面键合度(可用载碳量指标)和羟基活性方面的差异。可调整方法提高方法的适应性,或者让厂商备存足够的同批次的适应方法的填料。
如果得到正确使用、维护和保养,寿命会很长。在使用保护柱的情况下,有寿命超过1万针的实例。如果不用保护柱,即便得到很好保养维护,一般能用到2000针左右就不错了。不过保护柱频繁换柱芯的成本也不低,而且使用装填不好的保护柱还会影响分离质量。
对硅胶基质 ,流动相p H值和样品清洁程度对寿命影响较大。个人认为高柱温对寿命有影响,但不大,即使方法要求70摄氏的柱温,也没发现有显著的寿命缩短。进针前样品处理得越干净, 寿命当然越长。但样品前处理也是需要花钱的,需要在前处理成本、 成本和数据质量之间进行权衡。

文章来源:分析与GMP
分析与GMP
展源
何发
相关文章
-

色谱峰裂分,前拖尾的诊断
2020-05-27
-
峰拖尾与峰前沿原因分析与解决方案
2022-03-30
-

色谱峰裂分和前拖尾的诊断
2020-05-27
-

诊断色谱峰裂分、前拖尾
2020-05-27
-

色谱峰裂分,前拖尾的诊断
2020-05-27
-

色谱峰裂分、前拖尾的诊断
2020-05-27
-

GC峰拖尾的原因及解决方法
2023-12-06
-
色谱峰裂分、前拖尾情形的诊断
2020-05-27
-

气相色谱峰拖尾的原因及解决方法
2021-10-26

加载更多