溶剂效应——如何消除色谱峰这个隐形“杀手”???
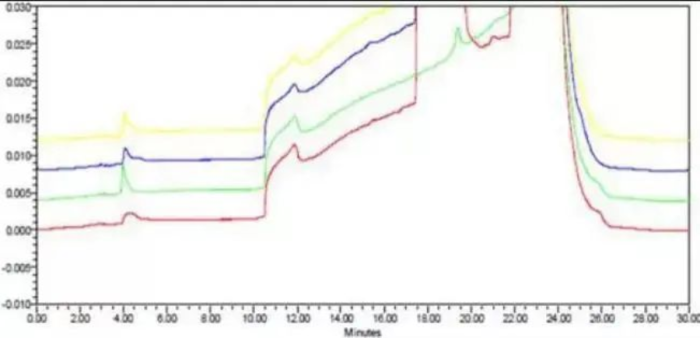
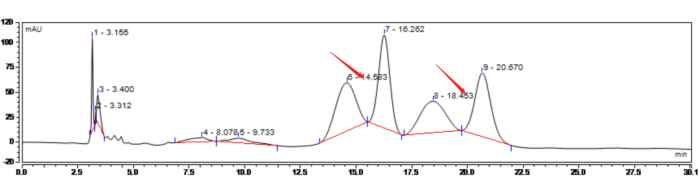
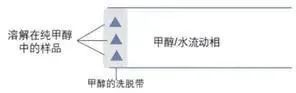
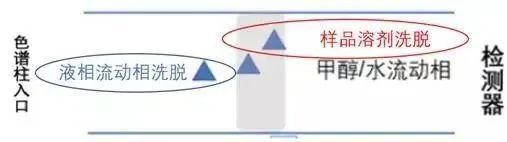

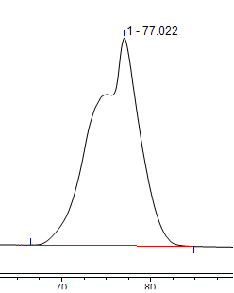
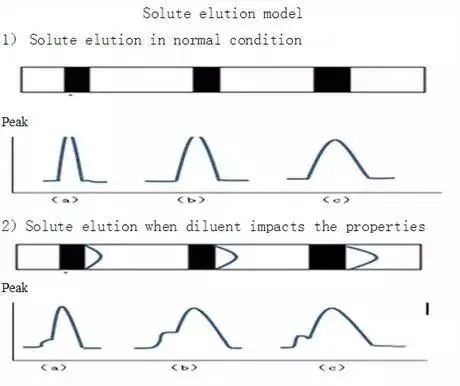
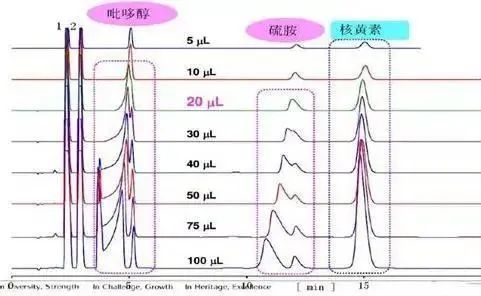
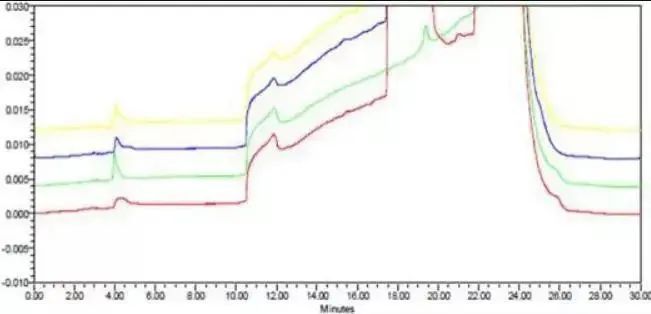
药物分析学社
展源
何发
相关文章
-

如何避免溶剂效应?
2021-07-25
-

【干货】如何避免溶剂效应
2022-02-09
-

溶剂效应是怎么回事?
2024-02-29
-

如何完美解决“溶剂效应”?
2022-08-04
-

如何完美解决“溶剂效应”?
2024-04-29
-

助焊剂的活性成分、溶剂等组成及研究进展
2020-05-27
-

液相分析过程中如何避免溶剂效应?
2023-04-18
-

【划重点】如何解决“溶剂效应”?
2025-03-19
-

反相色谱中溶剂效应的原理及解决方法
2023-02-20
-

液相分析过程中“溶剂效应”的那些事儿~
2023-06-16

加载更多