【解密】化药杂质研究分析技巧

杂质的来源分析
杂质的来源分析
对药物杂质研究时引入“质量源于设计( Quality byDesign,QbD)”的理念,可在药物生产之前根据具体工艺的合成机制、起始物料及各中间体的基本结构,初步勾画出产品的杂质谱。
杂质来源分析是制定药物杂质控制策略的基础,尤其是在对毒性杂质来源分析时,应分析所有合成和生产工艺中的试剂、中间体、副产物,推测可能产生的潜在杂质以及分析实际存在的杂质。在原料药合成结束后,药物的活性化合物虽然经过毒性分析已不含有“警示结构”(alerting structure),但是在生产过程中使用到含有警示结构的化合物则还需考虑其遗传毒性。
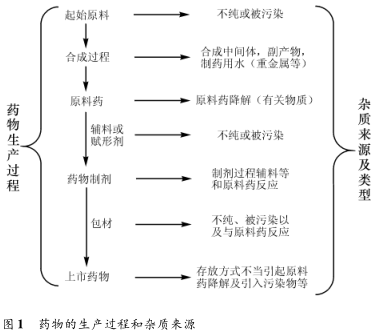
1、杂质的分类和存在的问题
①按理化性质分类:有机杂质、无机杂质及残留溶剂。
②按来源分类:工艺杂质( 包括合成中未反应完全的反应物及试剂、中间体、副产物等)、降解产物、从反应物及试剂中混入的杂质等。
③按毒性分类:毒性杂质和普通杂质等。
④按化学结构分类:其它甾体、其它生物碱、几何异构体、光学异构体和聚合物等。杂质的存在对药物的安全性和有效性有很大的影响,如何制定杂质的限度也是药物研发过程中一个非常敏感的问题。目前国内在药物杂质研究中存在的主要问题是“杂质限度制订的依据不充分”。研制单位通常是根据多批样品的研究结果确定,或凭所谓的“经验”制订。特别是方法学研究不全面,没有对杂质进行有效地分析,并进行定性研究,导致杂质研究不完善,成品与国外产品的质量差别较大。
2、一般要求
药品的开发需遵循三个原则:安全、有效和质量可控。因此,在制订质量标准中杂质的限度时,首先应从安全性和有效性方面考虑,尤其对具有药理活性或毒性的杂质,要严格控制其限度。其次,应考虑生产的可行性及批与批之间的正常波动,以及药物本身的稳定性。在保证安全和有效的前提下,根据工艺的波动情况制定合理的限度。此外,在药物研发过程中不同药物或者同一药物在不同研究阶段的侧重点是不一样的。因此,其杂质研究也各具特点,需要根据这些具体情况分别对待。
通常,可以根据药物规定的剂量确定降解产物的报告阈值、鉴定阈值和界定阈值,以此来确定杂质的限度。另外,亦可通过已知杂质的毒性数据,或其毒性研究结果来确定杂质的限度。
如,按ICH 要求,对于新的原料药,每日最大剂量大于2 g 时,单个杂质的界定阈值为0.05% ;每日最大剂量不大于2 g 时杂质的界定阈值0.1%或1 mg( 按严格的要求定)。当一个原料药的每日最大剂量为1.5 g 时,如按界定阈值0.1%计,则杂质的绝对摄入量为1.5 g×0.1% =1.5 mg,大于界定阈值1 mg ;
如按阈值1 mg 计,则杂质的限度值为1 mg÷1.5 g×100% =0.07%,与界定阈值(0.1% )不一致。在这种情况下,制订杂质限度的原则是“按严格的要求定”,即杂质限度应订为0.07%。
当然ICH 只是一般的要求,对于特殊杂质( 具有一定活性) 或杂质限度超出ICH 的要求时,应进行相关的安全性研究或根据文献数据,以判断限度的合理性。
对于已知杂质化合物,可参考相关的文献,或一些数据库( 如www.discoveryGate.com 网站) 查得其毒性数据,并根据药品临床拟用的剂量推算药物的安全限度。
应不高于0.05 mg/kg。若人体最大用药剂量为2 g时,产品中如含有该杂质,按人体重60 kg 计,其限度值为0.05 mg/kg×60 kg÷2 g×100% =0.15%。
一般情况可采用以下方法:
①对单个杂质进行毒理学研究,确定其NOAEL,制订该杂质的限度如,某创新药临床前试验样品中杂质A 的限度为0.34%,每天的最大用量拟定为10 mg。其临床研究用药,申报单位将杂质A 限度确定为0.5%。这一限度不但超过了ICH Q3 规定的常规杂质限量标准( 0.1% ),也超过了临床前试验样品中杂质A0.34%的限度。因此,在该品种的审评中,药审中心建议提供该杂质限度确定的依据。
研究者对此进行了说明,其推算过程如下:该药在大鼠重复给药6 个月的毒性试验中,无毒性反应剂量为26.5 mg·kg-1·d-1,则杂质A 的实际用量为26.5×0.34% =0.09 mg·kg-1·d-1。通过动物类比法转换为0.09 mg·kg-1·d-1×6=0.54 mg·/m-2·d-1( 大鼠在人体中mg/kg 剂量的转换因子为6),再用FDA 转换标准( 人体中mg/kg 与mg/m2 的转换因子为37),得出相当于60 kg 的人体耐受杂质A 的量为0.54×60÷37=0.88 mg/d。
考虑到该新药在临床研究中的最大给药剂量为10 mg,若按0.5%的杂质含量计算,每日的杂质A 最大剂量为0.05 mg/d,仅为杂质A无毒性反应剂量0.88 mg/d 的1/17,因此杂质A 的限度订为0.5%是可行的。
②根据毒理学实验样品杂质含量情况进行推算,制订出杂质的限度如,某个药物口服给药的每日最大剂量为60mg,相当于一个体重60 kg 的人给药剂量为1mg/kg。在13 周犬的口服给药毒理学研究中,确定其NOAEL 为2 mg/kg。
根据该批样品中杂质A 的含量为0.1%计算,杂质A 最大安全性限度为2 mg/kg×0.1% =2 μg/kg,换算到人体的杂质A 安全剂量约为1μg/kg( 犬在人体中mg/kg 剂量的转换因子为1.8),则杂质A 的限度为1 μg/kg÷1 mg/kg×100%=0.1%。

分析方法
分析方法的选择直接关系到杂质测定结果的专属性与准确性,因此,在进行杂质研究时首要问题是选择合适的杂质分析方法。
1、分析方法的选择
(1)有机杂质的分析方法
有机杂质的检测方法包括化学法、光谱法、色谱法等,因药物结构及降解产物的不同采用不同的检测方法。通过合适的分析技术将不同结构的杂质进行分离、检测,从而达到对杂质的有效控制。随着分离、检测技术的发展与更新,高效、快速的分离技术与灵敏、稳定、准确、适用的检测手段相结合,几乎所有的有机杂质均能在合适的条件下得到很好的分离与检测。
在质量标准中,目前普遍采用的杂质检测方法主要为高效液相色谱法(High Performance Liquid Chromatography;HPLC)、薄层色谱法(Thin Layer Chromatography;TLC)、气相色谱法(Gas Chromatography;GC)和毛细管电泳法(Capillary Electrophoresis;CE)。应根据药物及杂质的理化性质、化学结构、杂质的控制要求等确定适宜的检测方法。
由于各种分析方法均具有一定的局限性,因此在进行杂质分析时,应注意不同原理的分析方法间的相互补充与验证,如HPLC与TLC及HPLC与CE的互相补充,反相HPLC系统与正相HPLC系统的相互补充,HPLC不同检测器检测结果的相互补充等。
(2)无机杂质的分析方法
无机杂质的产生主要与生产工艺过程有关。由于许多无机杂质直接影响药品的稳定性,并可反映生产工艺本身的情况,了解药品中无机杂质的情况对评价药品生产工艺的状况有重要意义。对于无机杂质,各国药典都收载了经典、简便而又行之有效的检测方法。
对于成熟生产工艺的仿制,可根据实际情况,采用药典收载的方法进行质量考察及控制。对于采用新生产工艺生产的新药,鼓励采用离子色谱法及电感耦合等离子发射光谱-质谱(ICP-MS)等分析技术,对产品中可能存在的各类无机杂质进行定性、定量分析,以便对其生产工艺进行合理评价,并为制定合理的质量标准提供依据。
通常情况下,不挥发性无机杂质采用炽灼残渣法进行检测。某些金属阳离子杂质(银、铅、汞、铜、镉、铋、锑、锡、砷、锌、钴与镍等)笼统地用重金属限度检查法进行控制。
因在药品生产中遇到铅的机会较多,且铅易积蓄中毒,故作为重金属的代表,以铅的限量表示重金属限度。如需对某种(些)特定金属离子或上述方法不能检测到的金属离子作限度要求,可采用专属性较强的原子吸收分光光度法或具有一定专属性的经典比色法(如采用药典已收载的铁盐、铵盐、硒等的检查法检测药品中微量铁盐、铵盐和硒等杂质)。
虽然重金属检查法可同时检测砷,但因其毒性大,且易带入产品中,故需采用灵敏度高、专属性强的砷盐检查法进行专项考察和控制,各国药典收载的方法已历经多年验证,行之有效,应加以引用。
由于硫酸根离子、氯离子、硫离子等多来源于生产中所用的干燥剂、催化剂或pH调节剂等,考察其在产品中的残留量,可反映产品纯度,故应采用药典中的经典方法进行检测。如生产中用到剧毒物(如氰化物等),须采用药典方法检测可能引入产品中的痕量残留物。
对于药典尚未收载的无机杂质(如磷酸盐、亚磷酸盐、铝离子、铬离子等)的检测,可根据其理化特性,采用具有一定专属性、灵敏度等的方法,如离子色谱法、原子吸收分光光度法、比色法等。
2、分析方法的验证
杂质检测方法的验证应参照相关的技术指导原则进行,重点在于专属性和灵敏度的验证。专属性系指在其它成分可能共存的情况下,采用的方法能准确测定出被测杂质的特性。检测限是反映分析方法灵敏度的一个重要指标,所用分析方法的检测限一定要符合质量标准中对杂质限度的要求,最低检测限不得大于该杂质的报告限度。
为验证杂质分析方法的专属性,对于原料药,可根据其合成工艺,采用各步反应的中间体(尤其是后几步反应的中间体)、立体异构体、粗品、重结晶母液等作为测试品进行系统适用性研究,考察产品中各杂质峰及主成分峰相互间的分离度是否符合要求,从而验证方法对工艺杂质的分离能力。
为了考察方法能否有效检测出原料药或制剂中的降解产物,还可根据药物的化学结构特点、制剂的处方与工艺、储存条件等选用合适的酸、碱、光、热、氧化反应等加速破坏性试验来验证分析方法的专属性,必要时可采用二极管阵列检测器、质谱检测器等检测峰的纯度。
因为在强制降解试验条件下产生的降解产物较药品货架期产生的降解产物复杂、未知杂质多,分离难度大,上述分析方法可有效地显示各色谱峰的纯度,以免因分离度不符合要求,导致分析结果的不准确。
如不具备检测峰纯度的试验条件,可通过适当调整流动相的组成或比例使各色谱峰的相对保留时间发生改变,用同一份经加速破坏试验的供试品溶液进样,然后比较流动相调整前后杂质峰的个数;也可采用TLC法比较同一份经加速破坏试验的供试品溶液在不同展开系统下的斑点个数及位置,以此佐证杂质分析方法的专属性。
强制降解试验对于未知杂质的分离度考察是非常必要的,其目的主要是提供关于杂质(特别是降解物)与主成分的分离情况、样品稳定性及降解途径等重要信息。在试验过程中,应注意破坏性试验要适度,应着重考察敏感条件。
如产品在一定条件下稳定,则无必要再提高条件的剧烈程度进行重复试验。破坏试验的程度暂无统一要求,一般以强力破坏后主成分的含量仍占绝大部分为宜。此时已产生了一定量的降解产物,与样品长期放置的降解情况相似,考察此情况下的分离度更具有实际意义。
要达到这种破坏程度,需要在研究过程中进行摸索,先通过初步试验了解样品对光、热、湿、酸、碱、氧化条件的基本稳定情况,然后进一步调整破坏性试验条件(如光照强度、酸碱浓度、破坏的时间、温度等),以得到能充分反映降解产物与主成分分离的结果和图谱。
另外,通过比较试验前后主峰面积的变化,还可粗略估算降解物对主成分的相对响应因子,了解样品在各种条件下的稳定性,为包装及贮藏条件的选择等提供信息。对于性质相对稳定的药品,如有充分的文献依据或试验数据,则可以免做强制降解试验。
3、有机杂质的定量方法
有机杂质的检测一般多采用HPLC法,有时也采用TLC、GC等其它方法。
如采用HPLC法,须采用峰面积法,具体定量方法有
①外标法(杂质对照品法)、
②加校正因子的主成分自身对照法、
③不加校正因子的主成分自身对照法、
④峰面积归一化法。
①法定量比较准确,采用时应对对照品进行评估和确认,并制订质量要求。
②法应对校正因子进行严格测定,仅适用于已知杂质的控制。
③法的前提是假定杂质与主成分的响应因子基本相同。一般情况下,如杂质与主成分的分子结构相似,其响应因子差别不会太大。
④法简便快捷,但因各杂质与主成分响应因子不一定相同、杂质量与主成分量不一定在同一线性范围内、仪器对微量杂质和常量主成分的积分精度及准确度不相同等因素,所以在质量标准中采用有一定的局限性。
有关物质中包括已知杂质和未知杂质。已知杂质对主成分的相对响应因子在0.9-1.1范围内[3]时,可以用主成分的自身对照法计算含量,超出0.9-1.1范围时,宜用杂质对照品法计算含量,也可用加校正因子的主成分自身对照法。理想的定量方法为已知杂质对照品法与未知杂质不加校正因子的主成分自身对照法两者的结合。研究人员可根据实际情况选用合适的定量方法。
在选择合适的分析方法时还应考虑生产能力及质量控制的可行性等技术因素。尽管在附件中规定的限度精确到小数点后第二位,但并不意味着在日常的生产质控中所用的分析方法也要如此精确。如经过必要的验证,也可采用薄层色谱法等分析方法。在研发过程中,如果分析方法有改变,则应进行方法改变前后所得分析结果的可比性研究。
对于TLC法,通常采用杂质对照品法和主成分自身对照法进行检控制,后者仅限于杂质斑点的颜色与主成分斑点颜色一致的情况下使用。

药物分析之家
展源
何发
相关文章
-

【收藏】最全的化药杂质分析技术!
2023-11-29
-

药品元素杂质控制研究
2021-03-23
-

药物遗传毒性杂质研究思路
2023-06-08
-

化学药杂质分析和检测专题
2023-10-24
-

药物元素杂质分析技术专题
2020-08-13
-

药品研发中如何确定杂质限度
2021-04-07
-

药品杂质限度测定的验证要求
2022-07-06
-

口服药中的元素杂质含量测定
2020-05-27
-

前沿应用∣流动相含离子对试剂的化药杂质质谱鉴定方法
2022-11-18

加载更多